Osteogenesis imperfecta (choroba krehkých kostí): príčiny, prejavy, liečba
Osteogenesis imperfecta – genetická porucha krehkých kostí: príčiny, príznaky, diagnostika a moderné možnosti liečby a podpory života.
Osteogenesis imperfecta je genetická porucha, často nazývaná choroba krehkých kostí. Postihuje najmä tvorbu a štruktúru kolagénu typu I, základnej zložky kostí, čo vedie k ich zvýšenej lámavosti. Keďže ide najčastejšie o chybu v génoch zodpovedných za kolagén (najčastejšie mutácie v génoch COL1A1 a COL1A2), ochorenie sa často dedí ako autozomálne dominantné — to znamená, že abnormálny gén môže zdediť potomok od jedného z rodičov. V praxi však existujú aj nové („de novo“) mutácie a zriedkavejšie aj autozomálne recesívne formy. OI postihuje kolagénovú časť kosti (kolagénová tyčinka), ktorá zabezpečuje pevnosť kostí. Chorobu prvýkrát popísal Vrolík v roku 1854. Výskyt je relatívne zriedkavý — odhady hovoria približne 1:15 000 až 1:20 000 živonarodených.
Galéria obrázkov
10 Obrázky







Príčiny
Hlavnou príčinou sú mutácie ovplyvňujúce tvorbu alebo kvalitu kolagénu typu I (predovšetkým génové zmeny v COL1A1 a COL1A2). Výsledkom je oslabená kostná matrix, krehkejšie kosti a zvýšená náchylnosť na zlomeniny. Typ dedičnosti je väčšinou autozomálne dominantný, avšak viete sa stretnúť s rôznymi genetickými mechanizmami vrátane de novo mutácií a recesívnych foriem.
Prejavy a klinické príznaky
- Časté a opakované zlomeniny pri relatívne malom traume.
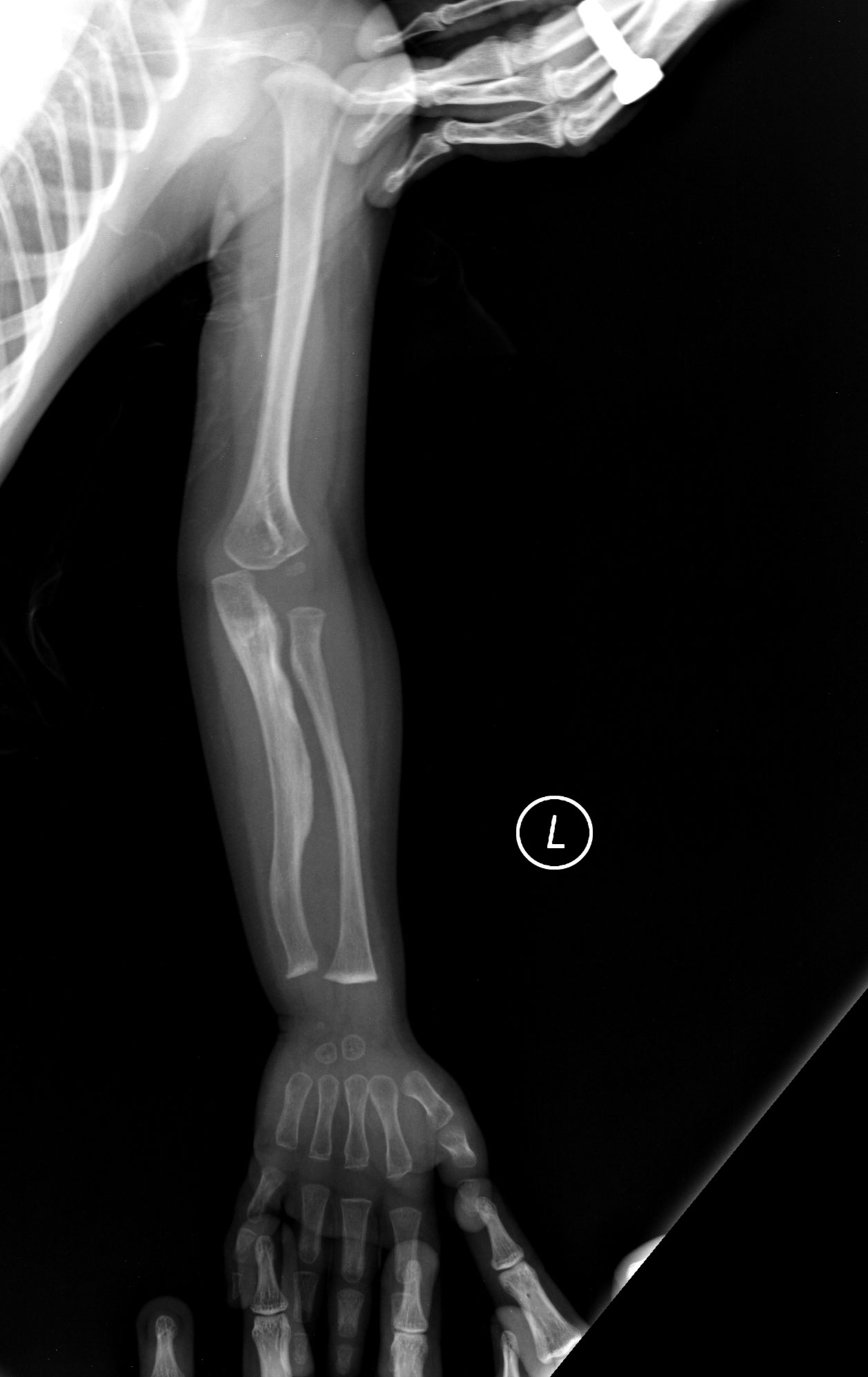
- Deformity kostry — skrátenie končatín, skolióza, tvarové zmeny dlhých kostí.
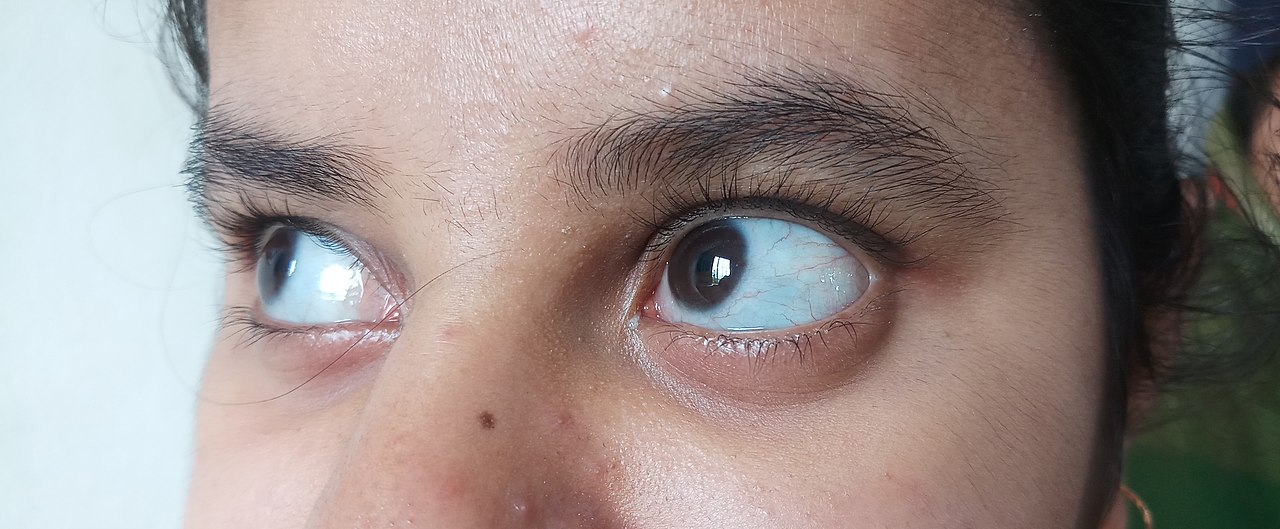
- Modré alebo fialové sfarbenie bielka oka (modré skléry), spôsobené tenšou spojivkou nad sklérou.
- Dentinogenesis imperfecta (krehké, abnormálne zuby) a zvýšená kazivosť.
- Strata sluchu — u približne 50 % pacientov sa môže objaviť v dospelosti (postihuje vonkajšie alebo stredné ucho alebo neurosenzoricky).
- Nižšia telesná výška, nadmerná pohyblivosť kĺbov, svalová slabosť.
- U ťažkých foriem — respiračné komplikácie a poruchy pľúcneho vývoja.
Stupne závažnosti (klasifikácia)
Najčastejšie používaná Sillence klasifikácia rozdeľuje OI na typy I–IV:
- Typ I – najčastejší a najľahší tvar, normálna alebo mierne znížená výška, modré skléry, časté zlomeniny v detstve, zvyčajne normálna až takmer normálna dĺžka života.
- Typ II – perinatálne letálny, ťažké kostné deformity a mnoho zlomenín už pri narodení.
- Typ III – progresívne deformujúca sa forma so značným počtom zlomenín, výrazným skracovaním a častou stratou sluchu; ťažká, ale život prežívaný do dospelosti v mnohých prípadoch.
- Typ IV – stredne ťažká forma, medzi typom I a III z hľadiska závažnosti.
Diagnostika
- Klinické vyšetrenie: anamnéza zlomenín, rodinná anamnéza, charakteristické znaky (modré skléry, zuby).
- Zobrazovacie vyšetrenia: röntgenové snímky ukážu typické zmeny kostí, deformity a liečené zlomeniny; DEXA sken môže hodnotiť kostnú hustotu.
- Laboratórne testy môžu pomôcť vylúčiť iné metabolické ochorenia kostí.
- Molekulárno-genetické testovanie: analýza mutácií v génoch COL1A1, COL1A2 a ďalších, ktoré potvrdí diagnózu, umožní presnejšiu prognózu a genetické poradenstvo.
- Prenatálna diagnostika: pri známej mutácii v rodine je možná molekulárna analýza embrya/plodu; ultrazvuk môže v 2. trimestri odhaliť ťažšie formy.
Liečba a manažment
Neexistuje liek, ktorý by OI úplne vyliečil, avšak dostupné terapie výrazne zlepšujú kvalitu života a znižujú počet zlomenín.
- Farmakologická liečba: Bisfosfonáty (napr. pamidronát u detí, zoledrónová kyselina) zvyšujú kostnú hustotu a môžu znižovať frekvenciu zlomenín. U dospelých sa v niektorých prípadoch skúša teriparatid; nové prístupy (denosumab) sú predmetom výskumu/užívania v špecifických situáciách.
- Ortopedické zákroky: intramedulárne rody (strieborné tyče) na vyrovnávanie a spevnenie dlhých kostí, korekčné operácie pri deformitách, stabilizácia zlomenín.
- Fyzioterapia a rehabilitácia: cvičenie na zlepšenie svalovej sily a rovnováhy, prevencia pádov, prispôsobené pohybové programy.
- Protézy a pomôcky: ortézy, chodítka, vozíky podľa potreby pre zlepšenie mobility a bezpečnosti.
- Stomatologická starostlivosť: ošetrenie dentinogenesis imperfecta, preventívna stomatológia a ochrana zubov.
- Hlasová a sluchová rehabilitácia: pravidelné vyšetrenia sluchu, nasadenie sluchových pomôcok alebo chirurgické riešenia pri indikácii.
- Výživa a suplementácia: dostatok vápnika a vitamínu D, udržiavanie optimálnej telesnej hmotnosti.
- Multidisciplinárna starostlivosť: koordinácia medzi ortopédmi, pediatrami, fyzioterapeutmi, genetikmi, stomatológmi a inými špecialistami pre komplexnú starostlivosť.
Prognóza
Prognóza závisí od typu OI. Miernejšie formy (typ I) umožňujú takmer normálnu dĺžku života s dobrou kvalitou života po adekvátnej liečbe a starostlivosti. Ťažké formy (typ II) sú perinatálne letálne. Typy III a IV majú variabilný priebeh s možnými chronickými obmedzeniami, deformitami a komplikáciami (respiračné problémy, chronická bolesť, strata sluchu).
Genetické poradenstvo a prevencia
Pre rodiny s diagnostikovanou mutáciou je dôležité genetické poradenstvo — vysvetlenie rizika dedenia, možností testovania pred a počas tehotenstva a diskusia o reprodukčných alternatívach. Pri podozrení na nový prípad sa odporúča genetické testovanie pacienta a potomkov.
Výskum a perspektívy
Prebieha výskum zameraný na nové liečebné prístupy vrátane pokusov s regeneratívnou medicínou, genetickými terapiami a novými liekmi, ktoré by mohli priamo ovplyvniť tvorbu kolagénu alebo zlepšiť kostnú kvalitu. Tieto terapie sú zatiaľ experimentálne alebo v klinických štúdiách.
Celková starostlivosť o ľudí s osteogenesis imperfecta kladie dôraz na prevenciu zlomenín, zlepšenie mobility, liečbu komplikácií a psychosociálnu podporu. Včasná diagnostika, pravidelné sledovanie a multidisciplinárny prístup výrazne zlepšujú dlhodobé výsledky.
Príznaky
Menej závažné príznaky OI môžu zahŕňať:
- ľahko zlomené kosti
- uvoľnené kĺby
- nízky svalový tonus
- modrá, fialová alebo sivá farba normálne bielej časti očí
- trojuholníkový tvar tváre
- sklon k vzniku skoliózy
- krehké zuby
OI má mnoho ďalších závažných a smrteľných príznakov vrátane dýchacích problémov a deformácie kostí.
Demografické údaje
OI sa vyskytuje rovnako u mužov aj u žien a môže postihnúť všetky etnické skupiny. OI sa vyskytuje v maternici a neexistuje na ňu liek. Zvyčajne sa zistí, keď si niekto zlomí veľa kostí; môže sa dať vyšetriť DNA na zistenie OI. Vyskytuje sa u 1 z 20 000 narodených detí.
Otázky a odpovede
Otázka: Čo je osteogenesis imperfecta?
Odpoveď: Osteogenesis imperfecta je genetická porucha, ktorá sa bežne nazýva choroba krehkých kostí. Oslabuje alebo ničí kolagénovú tyčinku, ktorá zabezpečuje pevnosť kostí, a vedie k tomu, že kosti sa častejšie lámu.
Otázka: Ako sa osteogenesis imperfecta dedí?
Odpoveď: Osteogenesis imperfecta je zvyčajne autozomálne dominantné ochorenie, čo znamená, že človek ho môže dostať, ak má abnormálny gén len jeden z jeho rodičov.
Otázka: Kto prvý identifikoval osteogenesis imperfecta?
Odpoveď: Vrolik ako prvý identifikoval osteogenesis imperfecta v roku 1849.
Otázka: Existuje liek na osteogenesis imperfecta?
Odpoveď: Bohužiaľ, osteogenesis imperfecta sa nedá vyliečiť.
Otázka: Aké sú štyri typy osteogenesis imperfecta?
Odpoveď: Štyri typy osteogenesis imperfecta sú typ jeden, typ dva, typ tri a typ štyri.
Otázka: Aké sú príznaky osteogenesis imperfecta tretieho typu?
Odpoveď: Ľudia s tretím typom osteogenesis imperfecta môžu mať viac ako 100 zlomenín pred pubertou. Ich oči majú často fialový, modrý alebo sivý odtieň a ľudia s týmto prípadom majú často aj poruchu sluchu.
Otázka: Ako častá je strata sluchu u ľudí s osteogenesis imperfecta?
Odpoveď: 50 % ľudí, ktorí majú osteogenesis imperfecta, má stratu sluchu v dospelosti.
Súvisiace články
Autor
AlegsaOnline.com Osteogenesis imperfecta (choroba krehkých kostí): príčiny, prejavy, liečba Leandro Alegsa
URL: https://sk.alegsaonline.com/art/73422
Zdroje
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"