Výpočtová chémia: definícia, metódy a aplikácie vo vývoji liekov a materiálov
Výpočtová chémia: definícia, metódy a aplikácie pri navrhovaní liekov a materiálov — predikcie štruktúr, vlastností a reaktivity pomocou moderných výpočtových metód.
Výpočtová chémia je odvetvie chémie, ktoré využíva počítačovú vedu na riešenie chemických problémov. Tieto programy počítajú štruktúry a vlastnosti molekúl a pevných látok. Výpočtová chémia zvyčajne dopĺňa informácie získané chemickými experimentmi. Dokáže predpovedať chemické javy, ktoré ešte neboli pozorované. Široko sa využíva pri navrhovaní nových liekov a materiálov.
Výpočtová chémia dokáže predpovedať štruktúru (t. j. očakávané polohy atómov molekuly), absolútne a relatívne (interakčné) energie, rozloženie elektronického náboja, dipóly a vyššie multipólové momenty, vibračné frekvencie, reaktivitu alebo iné spektroskopické veličiny a prierezy pre zrážky s inými časticami.
Výpočtová chémia sa zaoberá statickými aj dynamickými systémami. Vo všetkých prípadoch platí, že s rastúcou veľkosťou skúmaného systému rastie aj čas počítača a ďalšie využívané zdroje (napríklad pamäť a diskový priestor). Týmto systémom môže byť jedna molekula, skupina molekúl alebo pevná látka. Metódy výpočtovej chémie sa pohybujú od veľmi presných až po veľmi približné. Vysoko presné metódy sú zvyčajne použiteľné len pre malé systémy.
Galéria obrázkov
8 Obrázky


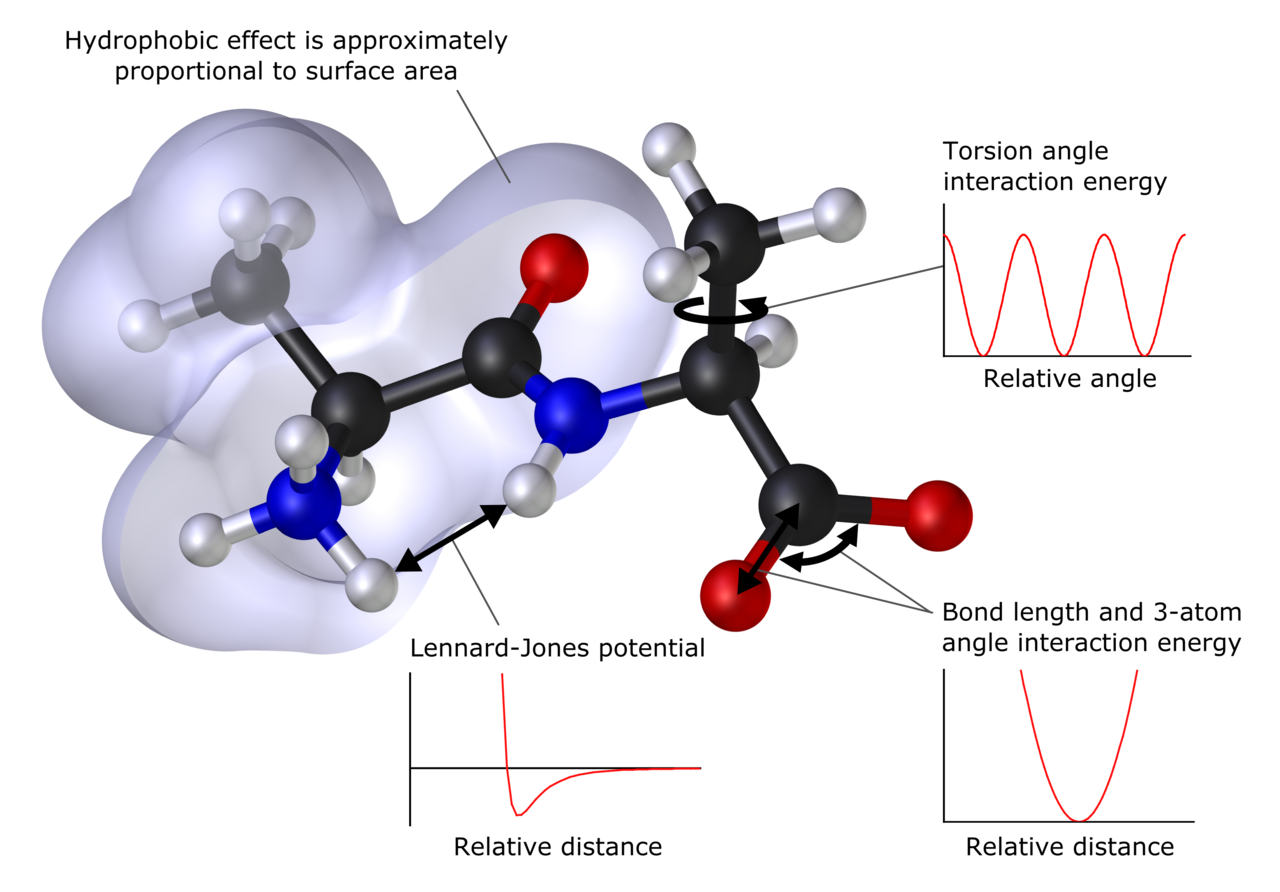


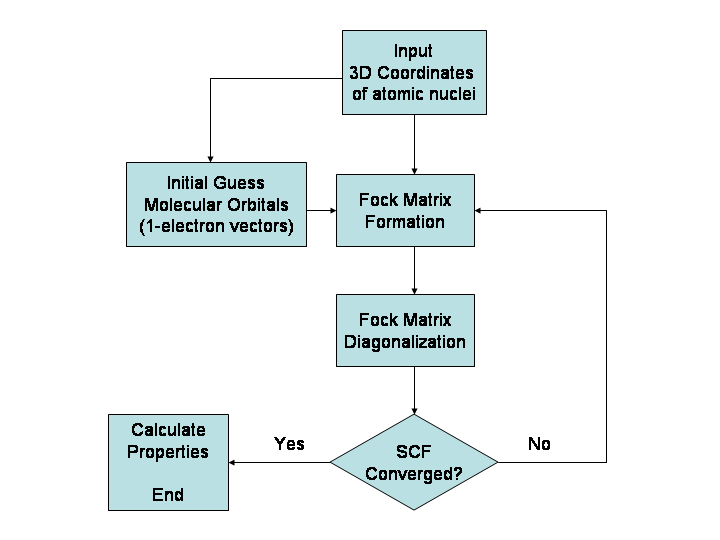
Hlavné metódy
- Ab initio (kvantovo-chemické) metódy: zahŕňajú Hartree–Fock (HF) a post-HF metódy (MP2, CCSD(T) a pod.). Sú teoreticky presné, ale rýchlosť rastie výrazne s veľkosťou systému, preto sa používajú najmä pre malé molekuly alebo ako referenčné metódy.
- Teória funkcionálu hustoty (DFT): veľmi rozšírená metóda vyvážajúca presnosť a výpočtové náklady; používa sa pre molekuly i pevné látky. Výber funkcionalu (B3LYP, PBE, ωB97X a pod.) a bázy výrazne ovplyvňuje výsledky.
- Semiempirické metódy: metódy ako PM3, PM6 alebo NDDO používajú zjednodušenia a experimentálne parametre; sú rýchlejšie, vhodné pre veľké systémy, ale menej presné.
- Metódy založené na silečných poliach (molekulárne mechaniky): používajú parametrizované potenciály (AMBER, CHARMM, OPLS) na popis pohybov atómov. Nepočíta sa explicitne elektronika, takže sú veľmi rýchle a vhodné na veľké systémy a dlhé dynamické simulácie.
- Molekulová dynamika (MD): simuluje pohyb atómov a molekúl v čase na základe síl z polí; slúži na štúdium dynamiky, stability komplexov, konformačných zmien a transportných procesov.
- Multiscale prístupy (QM/MM): kombinujú kvantovo-chemický opis reaktívnej časti systému (QM) s mechanickým opisom prostredia (MM), čo umožňuje študovať chemické reakcie v zložitejších prostrediach (enzýmy, katalyzátory).
- Monte Carlo (MC): metódy pre vzorkovanie konformačného priestoru a pri výpočtoch termodynamických vlastností.
Aplikácie vo vývoji liekov
- Virtuálne získavanie (virtual screening): prehľadávanie veľkých knižníc zlúčenín na identifikáciu kandidátov s vysokou afinitou k cieľovej biomolekule.
- Docking a hodnotenie väzby: predpovedanie spôsobu väzby ligand–cieľ a odhad relatívnej afinity.
- Výpočty voľnej energie viazania: metódy ako FEP, TI alebo MM-PBSA odhadujú presnejšie zmeny voľnej energie pri navrhovaní a optimalizácii ligandov.
- Modelovanie farmakokinetiky a toxicity: predikcie ADMET vlastností pomocou kombinácie fyzikálno-chemických výpočtov a strojového učenia.
- Podpora experimentov: návrh mutácií cieľových proteínov, vysvetlenie pozorovaných výsledkov a návrh ďalších experimentov.
Aplikácie v materiáloch
- Navrhovanie katalyzátorov: štúdium aktivačných energií, mechanizmov reakcií a identifikácia aktívnych centier.
- Bateriové a elektrochemické materiály: predpoveď transportu iónov, stability elektrolytov a rozhraní elektrod/elektrolytov.
- Polyméry a kompozity: simulácie mechanických vlastností, tepelnej stability a mikroskopickej morfológie.
- Fotovoltaika a optické materiály: predikcia absorpčných vlastností, energetických pásov a excitonových procesov.
- 2D materiály a povrchové vedy: štúdium štruktúry, elektronických vlastností a adsorpcie molekúl na povrchoch.
Praktické úvahy a obmedzenia
- Škálovanie náročnosti: výpočtové náklady rastú s počtom atómov; niektoré metódy (napr. CCSD(T)) môžu rásť ako vysoké mocniny veľkosti systému, zatiaľ čo MD s klasickou mechanikou škáluje priaznivejšie.
- Volba bázy a parametrov: kvalita výsledkov závisí od zvolenej bázy (STO-3G, 6-31G*, cc-pVDZ a pod.) a parametrizácie funkcionálov či silačných polí.
- Solvent a prostredie: modely rozpúšťadla (implicitné PCM/COSMO alebo explicitné molekuly) ovplyvňujú výsledky, najmä pre polárne interakcie a reaktívne mechanizmy.
- Vzorkovanie konformačného priestoru: pre veľké alebo flexibilné systémy môže byť hlavnou výzvou dosiahnuť dostatočné vzorkovanie relevantných konformácií.
- Validácia voči experimentu: výpočty často slúžia ako doplnok experimentov — treba ich overiť meraniami (spektroskopia, kalorimetria, difrakcia a pod.).
Softvér a hardvér
Existuje široké spektrum softvérových balíkov pre rôzne metódy: komerčné (napr. Gaussian) aj bezplatné/otvorené (napr. ORCA, NWChem, Quantum ESPRESSO, VASP, CP2K, GROMACS, LAMMPS). Výkon výpočtov sa zlepšuje nasadením výpočtov na klastroch, superpočítačoch a GPU akcelerovaných architektúrach; cloudové služby tiež umožňujú škálovanie pre high-throughput štúdie.
Súčasné trendy a výzvy
- Strojové učenie a dátovo riadené metódy: používa sa na rýchle predikcie vlastností, zlepšenie silačných polí a urýchlenie výpočtov bez výraznej straty presnosti.
- Enhanced sampling a multiskálové prístupy: metódy na lepšie vzorkovanie zložitých systémov (metadynamics, umbrella sampling) a kombinácie QM/MM pre reaktívne prostredia.
- Automatizácia a high-throughput: pracovné toky pre rýchle hodnotenie veľkých množín materiálov alebo ligandov.
- Presnosť vs. náklady: neustála snaha o metódy, ktoré ponúknu lepšiu presnosť pri prijateľných výpočtových nákladoch.
Zhrnutie: Výpočtová chémia poskytuje súbor nástrojov na pochopenie a predpovedanie chemických javov, ktoré dopĺňajú experimenty a urýchľujú vývoj liekov a materiálov. Správny výber metódy, validácia voči experimentu a starostlivé riešenie praktických otázok (vzorkovanie, solvent, parametre) sú kľúčové pre spoľahlivé výsledky.

Súvisiace stránky
- Bioinformatika
- Štatistická mechanika
Otázky a odpovede
Otázka: Čo je to výpočtová chémia?
Odpoveď: Výpočtová chémia je odvetvie chémie, ktoré využíva informatiku na riešenie chemických problémov. Môže sa použiť na výpočet štruktúry a vlastností molekúl a pevných látok, na predpovedanie chemických javov, ktoré ešte neboli pozorované, a na navrhovanie nových liekov a materiálov.
Otázka: Aké typy systémov skúma počítačová chémia?
Odpoveď: Výpočtová chémia skúma statické aj dynamické systémy. Systémom môže byť jedna molekula, skupina molekúl alebo pevná látka.
Otázka: Aké typy informácií môže poskytnúť počítačová chémia?
Odpoveď: Výpočtová chémia môže poskytnúť informácie, ako je štruktúra (pozície atómov), absolútne a relatívne energie, rozdelenie elektronických nábojov, dipóly a vyššie multipólové momenty, vibračné frekvencie, reaktivita alebo iné spektroskopické veličiny a prierezy pre zrážky s inými časticami.
Otázka: Aké presné sú metódy používané vo výpočtovej chémii?
Odpoveď: Presnosť metód používaných vo výpočtovej chémii sa pohybuje od veľmi presných až po veľmi približné. Vysoko presné metódy sú zvyčajne použiteľné len pre malé systémy.
Otázka: Ako počítačová chémia dopĺňa experimentálne údaje?
Odpoveď: Výpočtová chémia zvyčajne dopĺňa informácie získané chemickými experimentmi. Môže sa použiť na predpovedanie výsledkov, ktoré ešte neboli experimentálne pozorované.
Otázka: Má veľkosť skúmaného systému vplyv na to, koľko počítačového času je potrebného?
Odpoveď: Áno - s rastúcou veľkosťou študovaného systému rastie aj množstvo počítačového času potrebného na analýzu, ako aj zdroje, ako je pamäť a diskový priestor potrebný na ukladanie.
Súvisiace články
Autor
AlegsaOnline.com Výpočtová chémia: definícia, metódy a aplikácie vo vývoji liekov a materiálov Leandro Alegsa
URL: https://sk.alegsaonline.com/art/22297